Chemistry and Biochemistry
Chen Lab
Research in the Chen Lab
From detergents to painkillers, from Retina Displays to Eco Boxes – the quality of modern human life is sustained by an endless supply of chemical compounds produced through organic synthesis. Organic synthesis involves rearranging the way atoms are connected to each other. Orchestrating this dance of atoms requires chemists to strike a fine balance between aggressiveness and precision, for even a seemingly trivial difference in the connection, orientation or spatial arrangement of atoms can mean the difference between a life-saving drug and a life-ruining poison. As such, efficient synthesis of organic compounds requires the development of processes that are selective for products with the desired atomic connection, orientation, as well as three-dimensional shape.
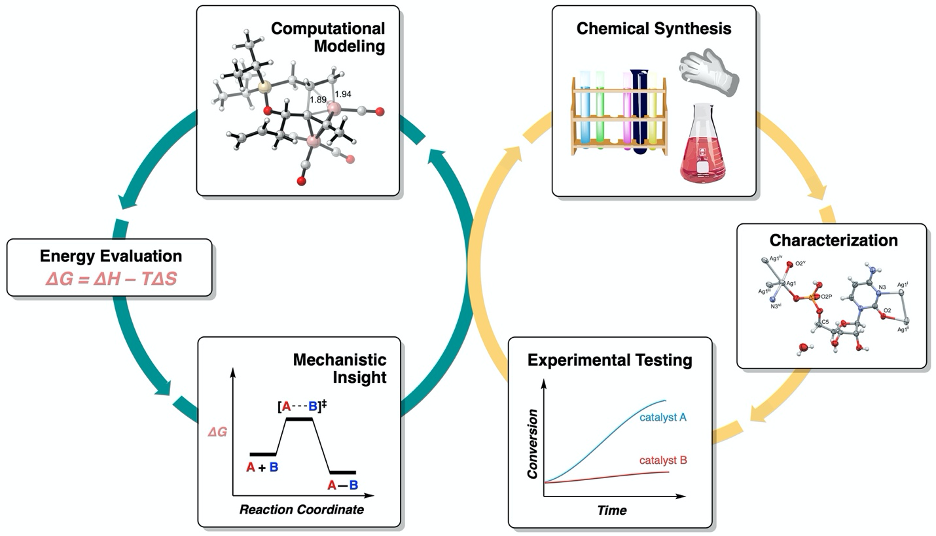
Despite the importance of chemical selectivity, the key to controlling it is often shrouded in mystery. Chemical reactions typically take place on time scales measured in femtoseconds (quadrillionths of a second), via even more fleeting structures dubbed transition states. While transition states are energetic bottlenecks that typically control the selectivity of reactions, they are exceedingly difficult to study by experimental means. Fortunately, modern computational chemistry has risen to the task and enabled chemists to obtain molecular-level insight into these transient but all-important structures. The overarching goal of my research is to leverage the synergy of computation and experiment to discover what drives chemical selectivity. The mechanistic insight we generate is in turn used to guide the design of reactions that are more selective, efficient and environmentally-friendly.
Students in my lab have the opportunity to learn the following experimental and computational techniques:
Experimental - organic and inorganic synthesis (including air- and water-free techniques such as glove boxes and Schlenk line usage), NMR spectroscopy (including heteronuclear NMR and specialized air-free NMR techniques), LCMS/GCMS, column chromatography, preparatory thin-layer chromatography, X-ray single crystal growth
Computational - Density Functional Theory (DFT) calculations (using Gaussian software), quasiclassical molecular dynamics (MD) simulations, reaction mechanism elucidation, automated conformational searches, Python scripting for data analysis (for students with more CS background)
Visit Prof. Chen's group website to learn more.